g. Run GROMACS MPI job
In this section, you will run a Gromacs MPI job distributed between two c5n.18xlarge nodes in your Kubernetes cluster.
1. Create MPIJob manifest
Execute the code block below to create a file named gromacs-mpi.yaml, containing an MPIJob manifest:
cat > ~/environment/gromacs-mpi.yaml << EOF
apiVersion: kubeflow.org/v2beta1
kind: MPIJob
metadata:
name: gromacs-mpi
namespace: gromacs
spec:
slotsPerWorker: 48
runPolicy:
cleanPodPolicy: Running
mpiReplicaSpecs:
Launcher:
replicas: 1
template:
spec:
restartPolicy: OnFailure
volumes:
- name: cache-volume
emptyDir:
medium: Memory
sizeLimit: 2048Mi
- name: data
persistentVolumeClaim:
claimName: fsx-pvc
initContainers:
- image: "${IMAGE_URI}"
name: init
command: ["sh", "-c", "cp /inputs/* /data; sleep 5"]
volumeMounts:
- name: data
mountPath: /data
containers:
- image: "${IMAGE_URI}"
imagePullPolicy: Always
name: gromacs-mpi-launcher
volumeMounts:
- name: cache-volume
mountPath: /dev/shm
- name: data
mountPath: /data
env:
- name: OMPI_MCA_verbose
value: "1"
command:
- /opt/view/bin/mpirun
- --allow-run-as-root
- --oversubscribe
- -x
- FI_LOG_LEVEL=warn
- -x
- FI_PROVIDER=shm,sockets
- -np
- "48"
- -npernode
- "48"
- --bind-to
- "core"
- /opt/view/bin/gmx_mpi
- mdrun
- -ntomp
- "1"
- -deffnm
- "/data/md_0_1"
- -s
- "/data/md_0_1.tpr"
Worker:
replicas: 1
template:
spec:
volumes:
- name: cache-volume
emptyDir:
medium: Memory
sizeLimit: 2048Mi
- name: data
persistentVolumeClaim:
claimName: fsx-pvc
containers:
- image: "${IMAGE_URI}"
imagePullPolicy: Always
name: gromacs-mpi-worker
volumeMounts:
- name: cache-volume
mountPath: /dev/shm
- name: data
mountPath: /data
resources:
limits:
memory: 8000Mi
requests:
memory: 8000Mi
EOF
2. Run the GROMACS MPIJob
To launch the GROMACS MPIJob, execute:
kubectl apply -f ~/environment/gromacs-mpi.yaml
Watch the pods in the gromacs namespace until the launcher pod enters Running state.
kubectl get pods -n gromacs -w
Press Ctrl-c to exit.
Follow the launcher logs while the pod is in Running state.
kubectl -n gromacs logs -f $(kubectl -n gromacs get pods | grep gromacs-mpi-launcher | head -n 1 | cut -d ' ' -f 1)
You should see GROMACS log entries similar to the ones shown below. Job output will hang on the line 50000 steps, 100.0 ps. while the simulation runs and will report the rest on simulation completion.
...
50000 steps, 100.0 ps.
Writing final coordinates.
Dynamic load balancing report:
DLB was off during the run due to low measured imbalance.
Average load imbalance: 24.7%.
The balanceable part of the MD step is 56%, load imbalance is computed from this.
Part of the total run time spent waiting due to load imbalance: 13.8%.
Average PME mesh/force load: 1.091
Part of the total run time spent waiting due to PP/PME imbalance: 4.6 %
NOTE: 13.8 % of the available CPU time was lost due to load imbalance
in the domain decomposition.
Dynamic load balancing was automatically disabled, but it might be beneficial to manually tuning it on (option -dlb on.)
You can also consider manually changing the decomposition (option -dd);
e.g. by using fewer domains along the box dimension in which there is
considerable inhomogeneity in the simulated system.
Core t (s) Wall t (s) (%)
Time: 2971.865 41.276 7199.9
(ns/day) (hour/ns)
Performance: 209.325 0.115
Press Ctrl-c to stop following the log.
While waiting for the GROMACS MPIJob to complete, explore the cluster utilization using either kubectl top node or the Grafana dashboards.
Example:
kubectl top node
Output:
NAME CPU(cores) CPU% MEMORY(bytes) MEMORY%
ip-192-168-86-187.ec2.internal 36114m 37% 5275Mi 1%
You should notice increased utilization of the cluster node cores. This is an indication that the MPI Job is running.
3. Delete MPIJob when completed
To delete the GROMACS MPIJob, after it is completed, execute:
kubectl delete -f ~/environment/gromacs-mpi.yaml
4. Check output files in FSx volume
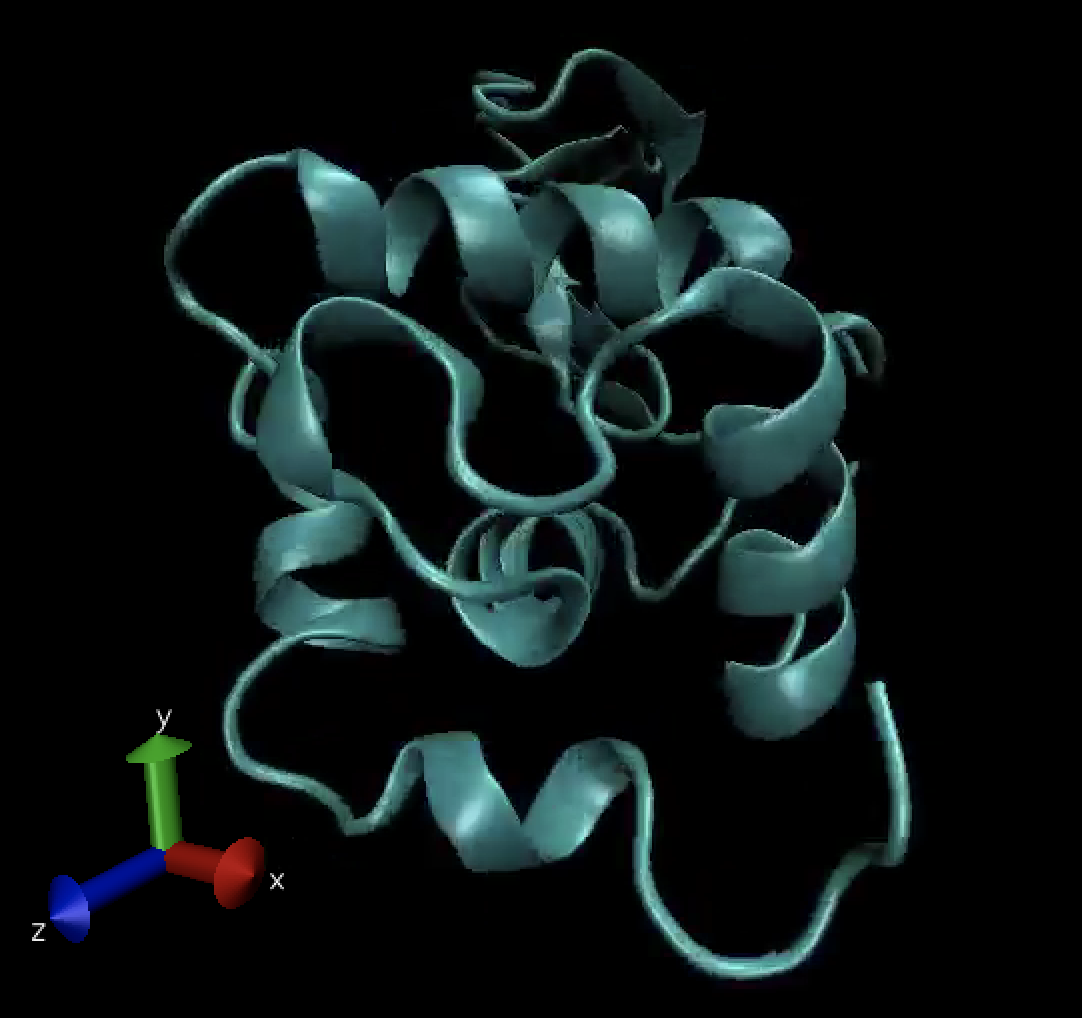
Congratulations, you have successfully run a tightly coupled MPI job on two nodes using GROMACS to simulate a protein (lysozyme) in a box of water with ions.
Once the job is completed the output files are in the FSx volume. To check that we will mount the volume in a new pod and open a shell in that pod.
Execute the code below to create a file named fsx-data.yaml, containing the pod manifest.
cat > ~/environment/fsx-data.yaml << EOF
apiVersion: v1
kind: Pod
metadata:
name: fsx-data
namespace: gromacs
spec:
containers:
- name: app
image: amazonlinux:2
command: ["/bin/sh"]
args: ["-c", "while true; do date; sleep 5; done"]
volumeMounts:
- name: data
mountPath: /data
volumes:
- name: data
persistentVolumeClaim:
claimName: fsx-pvc
EOF
Create the pod.
kubectl apply -f ~/environment/fsx-data.yaml
Check the pod status.
kubectl -n gromacs get pods
Once the pod is in Running state, open a shell into it.
kubectl -n gromacs exec -it $(kubectl -n gromacs get pods | grep fsx-data | head -n 1 | cut -d ' ' -f 1) -- bash
Describe the volumes mounted in the pod
df -h
Output:
Filesystem Size Used Avail Use% Mounted on
overlay 30G 6.7G 24G 23% /
tmpfs 64M 0 64M 0% /dev
tmpfs 185G 0 185G 0% /sys/fs/cgroup
192.168.106.172@tcp:/aqbp5bmv 1.1T 16M 1.1T 1% /data
/dev/nvme0n1p1 30G 6.7G 24G 23% /etc/hosts
shm 64M 0 64M 0% /dev/shm
tmpfs 185G 12K 185G 1% /run/secrets/kubernetes.io/serviceaccount
tmpfs 185G 0 185G 0% /proc/acpi
tmpfs 185G 0 185G 0% /sys/firmware
Notice the FSx volume is mounted under /data.
Check that the output data from the Gromacs MPI job is in the /data directory.
ls -alh /data
You should see a list of files like the one below:
...
total 9.4M
drwxr-xr-x 3 root root 33K Sep 30 04:29 .
drwxr-xr-x 1 root root 29 Sep 30 04:34 ..
-rw-r--r-- 1 root root 797K Sep 30 04:29 md_0_1.cpt
-rw-r--r-- 1 root root 8.2K Sep 30 04:29 md_0_1.edr
-rw-r--r-- 1 root root 2.3M Sep 30 04:29 md_0_1.gro
-rw-r--r-- 1 root root 36K Sep 30 04:29 md_0_1.log
-rw-r--r-- 1 root root 1.3M Sep 30 04:29 md_0_1.xtc
These are the files that the GROMACS simulations produced:
md_0_1.logcontains the GROMACS run output logs (open it up and inspect).md_0_1.grocontains the encoded protein structure.md_0_1.xtccontains particle trajectory information.md_0_1.edrcontains information about physical quantities, like energy, temperature, and pressure.md_0_1*.cptcontains checkpoint/restore data (can be used to resume the simulation).
When you are done inspecting the data files, exit the data pod shell.
exit
If the output data is copied to a linux desktop, it can be visualized with the VMD tool by using the following command (not covered here):
/usr/local/bin/vmd md_0_1.gro md_0_1.xtc
Visualized, the output looks like the image below and can be displayed as a movie over time: